November 13, 2024 report
This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
MassiveFold: Customizable version of AlphaFold reduces protein structure prediction time from months to hours
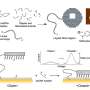
 and parameter options for AFmassive or ColabFold. MassiveFold then runs the alignments on a CPU, producing multiple sequence alignments (MSAs) and divides the structure predictions for massive sampling in batches to be run on GPUs. After completion, MassiveFold automatically gathers all predictions, ranks them following the AlphaFold ranking confidence score, the predicted template modeling score (pTM) and interface predicted template modeling score (ipTM), and generates plots. Credit: Nature Computational Science (2024). DOI: 10.1038/s43588-024-00714-4")
Scientists from Université de Lille, France, Linköping University, Sweden, and collaborating institutions have introduced MassiveFold, a new version of AlphaFold that dramatically reduces computing time for protein structure predictions from months to hours.
Protein structural prediction space is in a golden era of advancement, thanks to AI and machine learning tools. Biotechnology research heavily relies on discovering the correct protein structure to perform the desired task, with implications for just about any industry that interacts with biotechnology, from food to pharmaceuticals, fashion to biofuel, laundry detergent to agriculture, and seemingly everything in between.
DeepMind's AlphaFold and the AlphaFold Protein Structure Database have been major contributors. Initially trained for single protein chains, AlphaFold has since gone beyond this, showing high levels of accuracy in modeling complex protein assemblies during the recent CASP15-CAPRI round of blind structure prediction.
CASP (Critical Assessment of Structure Prediction) and CAPRI (Critical Assessment of Predicted Interactions) are two blinded benchmarks for protein prediction models to test their accuracy. Classically solved protein structures are chosen, and prediction tools are only given the amino acid sequences to work with. The closer a prediction model folding is to the actual structure, the higher the score.
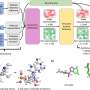
In a study titled "MassiveFold: unveiling AlphaFold's hidden potential with optimized and parallelized massive sampling," published in Nature Computational Science, the team introduces MassiveFold, an optimized and customizable version of AlphaFold that significantly enhances protein structure prediction capabilities.
Comparative analyses showed that MassiveFold could produce good models for several CASP15 targets, sometimes outperforming the recently published AlphaFold3. Depending on the target, either MassiveFold or AlphaFold3 produced the best models, suggesting tradeoffs in prediction strategies. In the future, these strategies are likely to be integrated.
MassiveFold significantly reduces computing time for protein structure predictions (from months to hours). This efficiency enables researchers to obtain results more rapidly, accelerating advancements in protein modeling and related scientific fields.
Previously, massive sampling within AlphaFold has been used to generate a large number of protein structure predictions to explore a wide range of possible conformations, which enhances the ability to model protein assemblies more accurately. These massive sampling tasks take intense computational resources beyond what many research teams have available.
MassiveFold addresses the challenges of high GPU resource demands and data storage that traditional AlphaFold applications face. Its ability to run predictions in parallel makes it practical even with limited computational resources.
MassiveFold is also scalable and customizable, capable of running on anything from a single computer to a large GPU infrastructure. This flexibility allows it to fully benefit from all available computing nodes, making it accessible to a wide range of research environments.
According to the study, the program is easy to use and install, requiring only a simple command line with a JSON parameter file. Its open-source availability to researchers encourages collaboration and further development within the scientific community, likely pushing the boundaries of what we can expect from clinical research and the biotech industry for many years to come.
The code for MassiveFold is publicly available on GitHub and Zenodo.
More information: Nessim Raouraoua et al, MassiveFold: unveiling AlphaFold's hidden potential with optimized and parallelized massive sampling, Nature Computational Science (2024). DOI: 10.1038/s43588-024-00714-4
Journal information: Nature Computational Science
© 2024 Science X Network